The ongoing COVID-19 pandemic is caused by SARS-CoV-2 coronavirus infection. The COVID-19 disease is air-borne, and usually causes fever/chills, cough/sore throat, shortness of breath/difficulty breathing, fatigue/tiredness, loss of taste or smell, headache, muscle ache/body ache and pains, sneezing/runny or congested nose, nausea/vomiting or diarrhoea. It is also fatal for some, while many also remain asymptomatic.
CSIR-CCMB and its two campuses, Medical Biotechnology Complex and Laboratory for the Conservation of Endangered Species (LaCONES) have been actively involved in fighting COVID-19 by studying the coronavirus as well as developing and enabling India-centric technologies.

ADVISORY ON FIGHTING CORONAVIRUS
Explore our Advisory page or Download the PDF

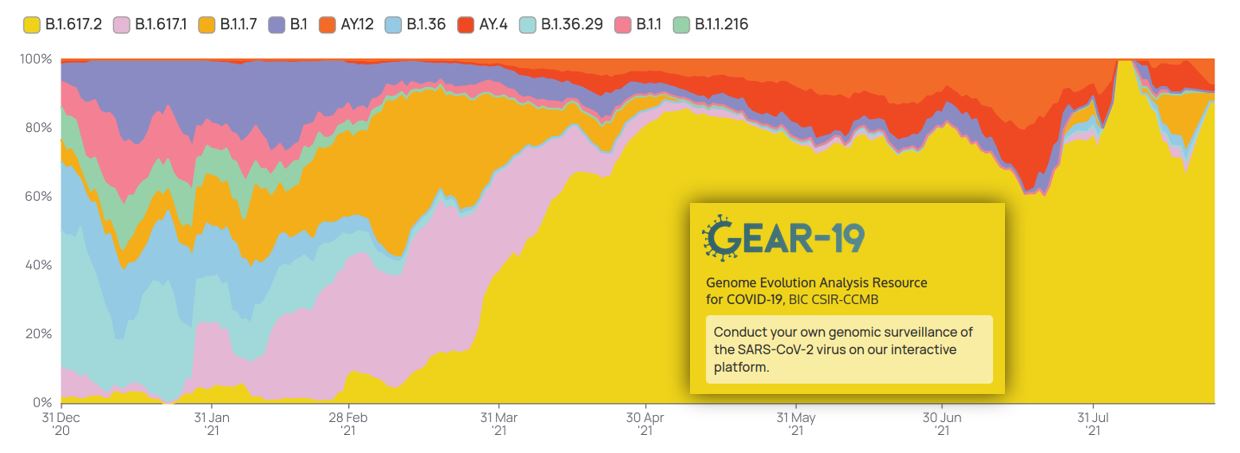
Genome Surveillance Work

COVID-19 CHRONICLES
To read the stories of our scientists involved in COVID-19 mitigation
COVID-19 NEWS & UPDATES

RESEARCH PROJECTS
The Centre for Cellular and Molecular Biology has been leading the research efforts in the war against the pandemic caused by the SARS-CoV-2 coronavirus.

